
1. Stoichiometry of metals in the ocean
The close relationship between the stoichiometry of nutrients dissolved in the upper ocean and the planktonic organisms that grow in these waters has long been recognized (1, 2). The stoichiometry of 106 C:16 N:1 P first summarized by Redfield has become a fundamental concept of marine biogeochemistry, with numerous studies using the ratio as a benchmark to assess ecosystem function. Decades after the work of Redfield, with the implementation of trace metal-clean techniques, oceanographers produced the first meaningful measurements of dissolved trace metals in the open ocean (3-5), and they found that many of the bioactive metals such as Fe, Zn, Ni, Cu and Cd are also depleted in surface waters and enriched at depth, similar to the macronutrients. Such nutrient-like behavior supported not only a growing understanding of the physiological roles that these metals play in phytoplankton physiology (6), but it also indicated that biological uptake and sub-surface remineralization were important processes for controlling the distributions of these bioactive metals in the ocean. Thus, the biogeochemical behavior of the micronutrient metals is in many ways analogous to that of the macronutrients N, P and Si.
In the open ocean far from coastal and shelf influences, dissolved concentrations of bioactive metals increase with depth at relatively consistent ratios to macronutrients (5), and these metal:nutrient remineralization ratios have been used to approximate the composition of sinking biogenic material and euphotic zone phytoplankton (7, 8). These ‘extended Redfield ratios’ have been compared to average compositions of marine phytoplankton species grown in culture (9-12), and the general agreement between these approaches further supports the importance of biological uptake and subsequent remineralization of trace metals in the upper ocean as key processes impacting trace metal geochemistry. Average metal:nutrient stoichiometries for phytoplankton have also been compared to dissolved stoichiometries in the ambient water, and relationships between these fractions have been used to estimate nutrient limitation and deficiency in the ocean (13). Thus, there is significant interest in controls on upper ocean metal stoichiometries, as well as the relationships between cellular/biological, particulate and dissolved fractions.
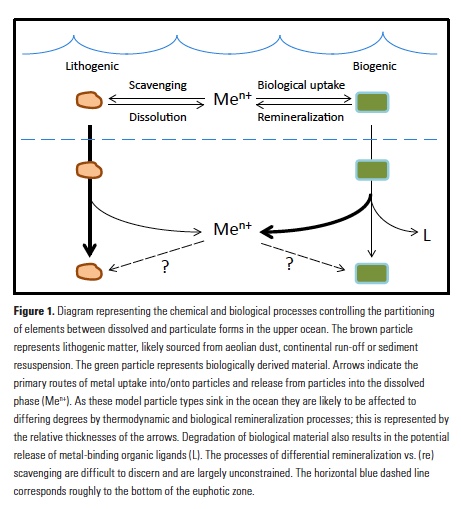
Analogous to macronutrients, there are also relationships between metal stoichiometries in phytoplankton and those in deeper waters of the ocean. Departures from these relationships are likely to provide insights into the internal biogeochemical cycling of metals in the ocean. Morel and Hudson (7) noted differences in the extended stoichiometries of plankton and the water column and concluded that they must reflect the relative efficiency of remineralization of the elements, as well as the propensity of elements to be scavenged onto sinking particles in the sub-surface ocean. Similarly, the rapid remineralization of trace metals from sinking plankton was addressed in seminal work by Collier and Edmond (14). Using carefully collected data on surface plankton material, and with more computational rigor than (7), they compared surface particle stoichiometries to deep water dissolved stoichiometries and calculated the relative remineralization of plankton-associated elements in sinking biogenic material. They noted significant differences among the behaviors of biogenic metals such as Cd, Ni and Fe due to their scavenging and remineralization behaviors. More recently, Morel (15) mused about these processes and their relationships to cellular biochemistry and evolution of phytoplankton physiology and ocean biogeochemistry.
Through the GEOTRACES program, the data to test and extend these early, relatively simple box models and stoichiometric comparisons are now available. Metal concentrations and stoichiometries for phytoplankton, bulk and size-fractionated particulate material, and co-located dissolved species have been measured in the North Atlantic and South Pacific Oceans thus far. Combined with data for non-bioactive metals such as Ti and Th, these data also provide the opportunity to discern the behavior and contributions of lithogenic vs. biogenic matter, as well as the processes of remineralization and scavenging.
2. Processes affecting dissolved and particulate stoichiometries of trace metals
Vertical profiles of dissolved macronutrients show characteristic depletion at the surface and enrichment at depth due to remineralization, and dissolved micronutrients often show the same behavior. However, the internal cycling of metals in the ocean is expected to differ from that of macronutrients for a few salient reasons. Some metals such as Fe are significantly less soluble than macronutrients and are prone to abiotic adsorption onto particulate surfaces (16). This process is driven by thermodynamics, and the accompanying process of desorption also occurs; the net observed process is typically called ‘scavenging’ (Fig. 1). Scavenging in the deep ocean causes concentrations of less soluble metals such as Fe and Al to decrease along the path of thermohaline circulation, in contrast to macronutrients and more soluble metals that may mimic macronutrient behavior such as Cd and Zn (17). In the absence of significant lateral nutrient inputs, the balance of scavenging and remineralization will influence the resulting vertical profiles of dissolved elements (18).
Another key difference between macronutrients and metals is the importance of abiotic particulate fractions such as lithogenic (e.g., aeolian dust and sediment) and authigenic (e.g., Fe- and Mn-oxyhydroxide) phases. While biogenic phases are almost universally produced at the surface and remineralized with depth, abiotic phases can exhibit very different and dynamic internal cycles (19). Dust events, lateral transport and poorly constrained scavenging processes can both deliver and remove specific metals alongside biological processes. Lithogenic phases are generally denser and more refractory than biogenic particles and detritus and are thought to sink more rapidly and remineralize more slowly and at greater depth (Fig. 1; 20). Lithogenic particles may also (re)scavenge metals differently than biogenic material. Efforts to examine these processes in sinking material have been extremely limited to date, with only a few studies examining metals in trace metal-clean sediment traps (21, 22). However, recently published datasets from the GEOTRACES program are shedding new light on the multiple facets of metal partitioning and how they affect subsurface remineralization and scavenging.
A comparison of metal:phosphorus ratios in the upper ocean illuminates some of these processes. Figure 2 displays Cd:P, Fe:P, Co:P and Ni:P ratios in particles in the upper 100m, 100-300m, and 300-1,000m of the water column in the middle of the North Atlantic basin. Particulate material is sub-divided into ratios for phytoplankton cells and non-lithogenic particles (corrected for lithogenic minerals using Ti; 19). Also plotted are dissolved remineralization ratios (that is, the slope of a linear regression between the dissolved metal and phosphate) for these upper ocean depth ranges. The close coupling of Cd and P biogeochemistry has long been recognized (4), and indeed we observe very close agreement (within a factor of about 2) between dissolved Cd:P remineralization and Cd:P in surface ocean particles, as well subsurface particles. Clearly these elements are remineralizing from sinking particles at similar rates. Such comparisons of particulate and dissolved constituents need to carefully consider the different residence times of these fractions and the likelihood for lateral inputs. Here, we have chosen to focus on stations from the mid-North Atlantic gyre, where the upper 700m of the water column consists primarily of a single water mass (23).
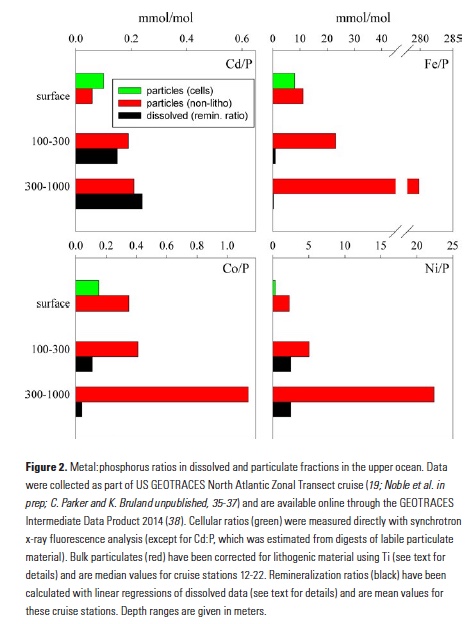
In contrast, the remineralization of Fe and P are quickly decoupled in the water column (Fig. 2). Between 100 and 300m, typically the depth of most rapid regeneration of sinking organic material, labile particulate Fe:P has more than doubled from that in surface waters, and the Fe:P ratio of remineralized dissolved elements (0.98 mmol/mol) is more than 10-fold below that of the labile material that is sinking into these waters. Looking deeper into the water column, Fe and P continue to decouple in labile (i.e., non-lithogenic) particulates, with Fe:P of 300-1,000m particles increasing 10-fold and the dissolved remineralization ratio being nearly 1,000-fold lower (0.35 mmol/mol). Additionally, organic ligands play an important role in stabilizing dissolved Fe (24), so dissolved Fe and P ratios may be further decoupled by biological processes impacting the production and fate of these ligands (20).
A strength of GEOTRACES datasets is their wide coverage of the periodic table, and additional insights can be gained from looking at the behaviors of other bioactive trace metals that are also incorporated into sinking biogenic material. Co:P ratios in particles and remineralized dissolved fractions in the water column follow the same trend as Fe, but the decoupling of Co and P is much more subtle than with Fe, presumably due to differences in ligand coordination and Co co-oxidation with Mn (25, 26). Dissolved Co:P remineralization ratios at 100-300m generally match those found in phytoplankton and drop only 3-fold below 300m. Similarly, labile particulate Co:P ratios don’t change between 0-100m and 100-300m, also indicating that Co and P remineralize in tandem in the upper 300m. Below 300m, labile particulate Co:P increases approximately 3-fold (in contrast with Fe:P, which increases 12-fold), and this depth effect matches the effect in dissolved remineralization ratios. Thus, even though Fe and Co are considered hybrid metals that display both biological uptake and scavenging, there are clear differences in the behaviors of these metals. Nickel provides yet another perspective on the coupling of metals and P. Dissolved remineralization ratios in both subsurface depth ranges closely resemble surface ocean labile particles, supporting the biological coupling of Ni and P (5). However, residual labile particulate Ni:P increases 2- to 4-fold in successive depth ranges, indicating that remineralization is rather decoupled. Given that Ni seems to be associated with both organic material and opal frustules in diatoms (27), it may be that Ni and P are remineralized from particulate organic matter in tandem, but some Ni remains associated with sinking biogenic silica in the ocean.
3. Additional tools to explore and differentiate remineralization processes
The GEOTRACES program has welcomed the application of new analytical approaches that further enable us to study the cycling of metals in the ocean. Spectroscopy and quantitative imaging methods using synchrotron radiation have become more common in the past decade (28), and these allow us to analytically distinguish the behaviors of different fractions of particle assemblages. During the FeCycle II project, a GEOTRACES process study, the fate of Fe was tracked during a spring diatom bloom (29). Diatom cells from the dominant bloom species (Asterionellopsis glacialis) were collected in surface waters and from trace-metal clean sediment traps at 100m and 200m in the 48h following the decline of the bloom. Synchrotron x-ray fluorescence (SXRF) analyses of individual cells showed that constituent elements were lost from sinking cells at notably different rates (Fig. 3). Phosphorus was rapidly released from sinking cells, with mean P quotas decreasing 55% and 73% from surface values by 100m and 200m, respectively (30). However, only 25% of cellular Fe was lost from cells sinking through the upper 200m, while 61% of cellular Ni was remineralized. This supports the story told by the bulk biogeochemical data from the North Atlantic: Ni is remineralized largely to a similar degree as P, while Fe is lost more slowly from sinking biogenic material.
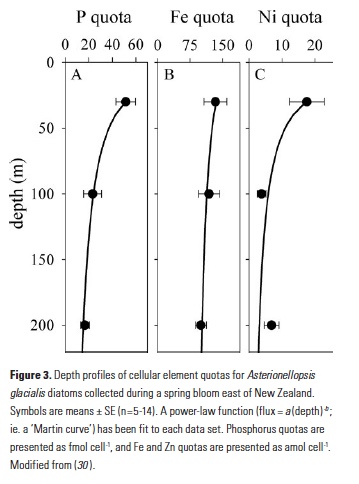
Application of microanalytical techniques such as SXRF can be combined with bulk approaches to further advance understanding of subsurface metal remineralization and cycling. In FeCycle II, Fe:P of sinking A. glacialis cells increased, on average, only 2.3-fold in the upper 200m, while Fe:P in bulk particulate matter increased more than 13-fold (30). This indicates that the behavior of sinking cells was not representative of the full particle assemblage. Iron and P were likely more completely decoupled in sinking fecal pellets and detrital material (which appears to have contributed significantly to the particulate Fe pool during FeCycle II; 31) than in intact sinking cells. Further application of this approach will allow us to not only distinguish between the behavior of biogenic and lithogenic fractions (Fig. 1), but potentially also between detrital particles. By considering metals such as Mn that are prone to oxidation and scavenging in the subsurface ocean (32, 33), it may also be possible to separate abiotic scavenging from net biological remineralization (Fig. 1). Additionally, 2D (and potentially 3D) mapping of elements within cells and particles also provides information about the spatial and potentially chemical associations of elements with particles (30, 34).
The GEOTRACES program is generating unprecedented data, both in terms of quality and quantity, regarding the cycling of bioactive trace metals in the ocean. Syntheses of these data, and integration of insights from novel microanalytical tools, as well as transcriptomic and proteomic approaches, are resulting in substantial advances in our understanding of metal biogeochemistry. No longer are we limited to a few painstakingly collected dissolved metal profiles. There is now painstakingly collected full-depth coverage of most ocean basins, including in many cases dissolved and particulate fractions of nearly all biogenic elements, enabling testing of early hypotheses about trace metal cycling and parameterization of these processes into next-generation ocean biogeochemical models.
Authors
Benjamin S. Twining, Daniel C. Ohnemus, Renee L. Torrie (Bigelow Laboratory for Ocean Sciences)
Acknowledgments
This work was funded by NSF grant OCE-1232814 to BST. RLT was funded by NSF REU grant 1460861 to Bigelow Laboratory for Ocean Sciences.
References
1. A. C. Redfield, in James Johnstone Memorial Volume, R. J. Daniel, Ed. (Liverpool University Press, 1934), pp. 176-192.
2. A. C. Redfield, Amer. Scientist 46, 205-221 (1958).
3. E. A. Boyle, J. M. Edmond, Nature 253, 107-109 (1975).
4. E. A. Boyle, F. Sclater, J. M. Edmond, Nature 263, 42-44 (1976).
5. K. W. Bruland, Earth Plan. Sci. Lett. 47, 176-198 (1980).
6. J. R. R. Frausto da Silva, R. J. P. Williams, The Biological Chemistry of the Elements: the inorganic chemistry of life. (Oxford University Press, Oxford, ed. 2nd, 2001), pp. 575.
7. F. M. M. Morel, R. J. M. Hudson, in Chemical Processes in Lakes, W. Stumm, Ed. (John Wiley & Sons, New York, 1985), pp. 251-281.
8. K. W. Bruland, J. R. Donat, D. A. Hutchins, Limnol. Oceanogr. 36, 1555-1577 (1991).
9. T. Y. Ho et al., J. Phycol. 39, 1145-1159 (2003).
10. W. G. Sunda, Marine Chem. 57, 169-172 (1997).
11. W. G. Sunda, S. A. Huntsman, Limnol. Oceanogr. 40, 132-137 (1995).
12. W. G. Sunda, S. A. Huntsman, Limnol. Oceanogr. 40, 1404-1417 (1995).
13. C. M. Moore et al., Nature Geosci. 6, 701-710 (2013).
14. R. Collier, J. Edmond, Prog. Oceanogr. 13, 113-199 (1984).
15. F. M. M. Morel, Geobiol. 6, 318-324 (2008).
16. M. Whitfield, D. R. Turner, in Aquatic Surface Chemistry: Chemical Processes at the Particle-Water Interface, W. Stumm, Ed. (John Wiley & Sons, Inc., 1987), pp. 457-493.
17. K. W. Bruland, M. C. Lohan, in The Oceans and Marine Geochemistry: Treatise on Geochemistry, H. Elderfield, Ed. (Elsevier, Oxford, 2003), pp. 23-47.
18. P. W. Boyd, M. J. Ellwood, Nature Geosci. 3, 675-682 (2010).
19. D. C. Ohnemus, P. J. Lam, Cycling of lithogenic marine particles in the US GEOTRACES North Atlantic Transect. Deep-Sea Res. II 116, 282-302 (2015).
20. P. W. Boyd et al., Limnol. Oceanogr. 55, 1271-1288 (2010).
21. R. D. Frew et al., Glob. Biogeochem. Cycles 20, GB1S93, doi:10.1029/2005GB002558 (2006).
22. C. H. Lamborg, K. O. Buesseler, P. J. Lam, Deep-Sea Res. II 55, 1564-1577 (2008).
23. W. J. Jenkins et al., Deep-Sea Res. II 116, 6-20 (2015).
24. M. Gledhill, K. N. Buck, Frontiers Microbiol. 3, doi: 10.3389/fmicb.2012.00069 (2012).
25. J. W. Moffett, J. Ho, Geochim. Cosmochim. Acta 60, 3415-3424 (1996).
26. A. E. Noble et al., Limnol. Oceanogr. 57, 989-1010 (2012).
27. B. S. Twining et al., Glob. Biogeochem. Cycles 26, GB4001, doi:4010.1029/2011GB004233 (2012).
28. P. J. Lam et al., Prog. Oceanogr. 133, 32-42 (2015).
29. P. W. Boyd et al., Geophys. Res. Lett. 39, doi:10.1029/2012GL053448 (2012).
30. B. S. Twining et al., Limnol. Oceanogr. 59, 689-704 (2014).
31. A. L. King et al., Biogeosci. 9, 667-687 (2012).
32. J. P. Cowen, K. W. Bruland, Deep-Sea Res. 32, 253-272 (1985).
33. D. C. Ohnemus et al., Limnol. Oceanogr. In press (2016).
34. J. Nuester, S. Vogt, B. S. Twining, J. Phycol. 48, 626-634 (2012). 35. B. S. Twining et al., Prog. Oceanogr. 137, 261-283 (2015).
36. M. Hatta et al., Deep-Sea Res. II 2015, 117-129 (2015).
37. S. Roshan, J. Wu, Glob. Biogeochem. Cycles 29, 2082-2094 (2015).
38. E. Mawji et al. Marine Chem. 177, 1-8 (2015).

