Coccolithophores and the carbon cycle
Increasing atmospheric CO2 concentrations are resulting in both warmer sea surface temperatures due to the greenhouse effect and increasingly carbon-rich surface waters. The ocean has absorbed roughly one third of anthropogenic carbon emissions (1), causing a shift in carbon chemistry equilibrium to more acidic conditions with lower calcium carbonate saturation states (ocean acidification). Organisms that produce calcium carbonate structures are thought to be particularly susceptible to these changes (2-4).
Coccolithophores are the most abundant type of calcifying unicellular micro-algae in the ocean, producing microscopic calcium carbonate plates called coccoliths (5). Low-pH conditions have been shown to disrupt the formation of coccoliths (calcification; e.g., (6)). Therefore, it is generally expected that a higher-CO2 ocean will cause a reduction in calcification rates or a decrease in the abundance of these calcifiers. Such changes could have far-reaching consequences for marine ecosystems, as well as global carbon cycling and carbon export to the deep sea.
Coccolithophores use sunlight to synthesize both organic carbon through photosynthesis and particulate inorganic carbon (PIC) through calcification. Detrital coccolithophore shells form aggregates with organic material, enhancing carbon export to the deep sea (7). Coccolithophores also produce dimethyl sulfide (DMS), a climatically relevant trace gas that impacts cloud formation, ultimately influencing Earth’s albedo (8, 9). At the ecosystem level, coccolithophores compete for nutrients with other phytoplankton and provide energy for the rest of the marine food web. Coccolithophores have a broad range of irradiance, temperature, and salinity tolerances (10, 11). Moreover, their relatively low nutrient requirements and slow growth rates offer a competitive advantage under projected global warming and ocean stratification (5). This plasticity and opportunistic behavior can be critical for persistence in a changing oceanic environment. Given the wide range of biogeochemical and ecological processes impacted by coccolithophores, it is important to assess how anthropogenic changes may affect coccolithophore growth and calcification.
Many laboratory studies have investigated the impact of future environmental conditions on coccolithophores by decreasing pH, increasing dissolved inorganic carbon, and increasing temperature to mimic end-of-century projections. However, these have often yielded conflicting results: Some show a decrease, while others show no change or even increased calcification (e.g., (6, 12, 13)). For example, laboratory simulations of contemporary oceanic changes (increasing CO2 and decreasing pH) show that coccolithophores have the ability to modulate organic carbon production and calcification in response to variable amounts of dissolved inorganic carbon (DIC) but that low pH only affects these processes below a certain threshold (14). Another study indicated that coccolithophores could adapt to warming and highCO2 levels over the course of a year, maintaining their relative particulate organic carbon (POC) and PIC production per cell (15). One of the limitations of all laboratory experiments is that only a handful of species (and strains) are studied, which is only a tiny fraction of the diversity present in the oceans. Given the challenges of extrapolating laboratory results to real world oceans, studying recent trends in natural populations may lead to important insights.
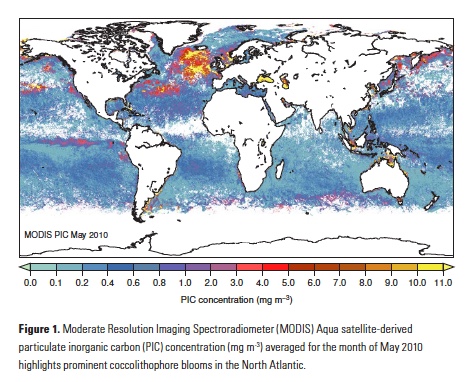
The North Atlantic is both a region with rapid accumulation of anthropogenic CO2 (1) and an important coccolithophore habitat (Fig. 1), making this region a good starting point to search for in situ evidence of anthropogenic carbon effects on diverse coccolithophore populations. Two recent studies did precisely that: Rivero- Calle et al. (2015)(16) in the subpolar North Atlantic, and Krumhardt et al. (2016)(17) in the North Atlantic subtropical gyre. Using independent datasets, these two studies concluded that coccolithophores in the North Atlantic appear to be increasing in abundance and, contrary to the prevailing paradigm, responding positively to the extra carbon in the upper mixed layer.
Evidence from long-term in situ monitoring (two independent case studies)
Rivero-Calle et al. (2015) used data from the Continuous Plankton Recorder (CPR), a filtering device installed on ships of opportunity, to assess changes in coccolithophore populations from 1965 to 2010 in the subpolar North Atlantic. This highly productive, temperate region is dominated by large phytoplankton and characterized by strong seasonal changes in the mixed layer depth, nutrient upwelling, and gas exchange that lead to intense, well-established spring phytoplankton blooms.
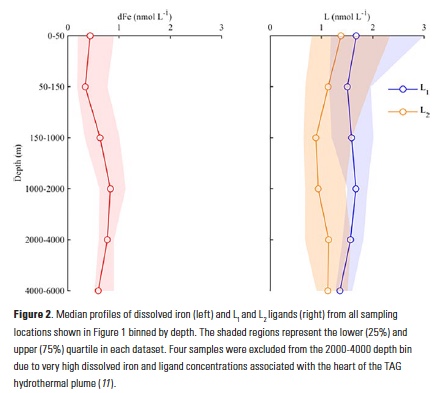
Because coccolithophore cells are smaller than the mesh size used by the CPR, they cannot be accurately quantified in the CPR data set. Some coccolithophore cells do, however, get caught in the mesh and their occurrence (i.e. probability of presence) can be calculated and serve as a proxy for coccolithophore abundance. Using recorded presence or absence of coccolithophores over this multidecadal time-series, the authors showed that coccolithophore occurrence in the subpolar North Atlantic increased from being present in only 1% of samples to > 20% over the past five decades (Fig. 2). To assess the importance of a wide range of diverse environmental drivers on changes in coccolithophore occurrence, Rivero-Calle and co-authors used random forest statistical models. Specifically, they examined more than 20 possible biological and physical predictors, including CO2 concentrations, nutrients, sea surface temperature and the Atlantic Multidecadal Oscillation (AMO), as well as possible predators and competitors. Global and local CO2 concentrations were shown to be the best predictors of coccolithophore occurrence. The AMO, which has been in a positive phase since the mid-1990s (Fig. 2) and is associated with anomalously warmer temperatures over the North Atlantic, was also a good predictor of coccolithophore occurrence, but not as strong of a predictor as CO2.
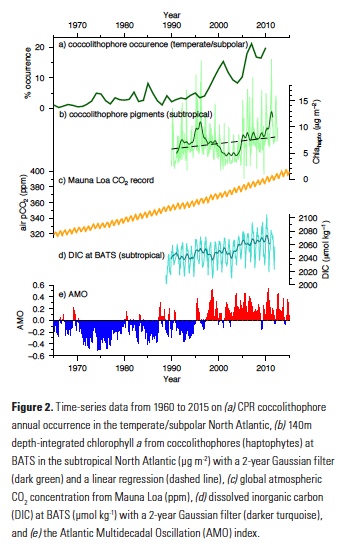
The authors hypothesize that the synergistic effects of increasing anthropogenic CO2, the recent positive phase of the AMO, and increasing global temperatures contributed to the observed increase in coccolithophore occurrence in the CPR samples from 1965 to 2010. Complementing the Rivero-Calle et al. (2015) study, Krumhardt et al. (2016) used phytoplankton pigment concentration data from the long-running Bermuda Atlantic Time-series Study (BATS) and satellite-derived PIC data to assess recent changes in coccolithophore abundance in the subtropical North Atlantic. This region of the North Atlantic is characterized by Ekman convergence and downwelling, resulting in an oligotrophic environment. Despite relatively low productivity, subtropical gyres cover vast expanses of the global ocean and are thus important on a global scale.
In the North Atlantic subtropical gyre, researchers at BATS have performed phytoplankton pigment analyses since the late 1980s, as well as a suite of other oceanographic measurements (nutrients, temperature, salinity, etc.). This rich dataset provided insight into phytoplankton dynamics occurring at BATS over the past two decades. Coccolithophores contain a suite of pigments distinctive to haptophytes. Though there are many species of non-calcifying haptophytes in the ocean (18), the main contributors to the haptophyte community in oligotrophic gyres are coccolithophores (19). Using a constant haptophyte pigment to chlorophyll a ratio Krumhardt et al. (2016) quantified relative abundance of the coccolithophore chlorophyll a (Chlahapto) over the BATS time-series. A simple linear regression revealed that coccolithophore pigments have increased in the upper euphotic zone by 37% from 1990 to 2012 (Figure 2). On the other hand, total chlorophyll a at BATS only increased slightly over this time period.
While satellite-derived chlorophyll a is used as a proxy for biomass and abundance of the entire phytoplankton community (20), satellite-derived PIC is formulated to specifically retrieve calcium carbonate from coccolithophore shells (21, 22). Therefore, satellite PIC can be used as a proxy for coccolithophore abundance. Although there has been virtually no change in total chlorophyll a over most of the North Atlantic subtropical gyre over the satellite era (1998-2014), predominantly positive trends were shown over this time period for PIC (17). This indicates that coccolithophore populations appear to be increasing over and above other phytoplankton species in the subtropical gyre.
Like Rivero-Calle et al., Krumhardt et al. explored possible environmental drivers of this increase in coccolithophore pigments at BATS and coccolithophore PIC throughout the gyre. They performed linear correlations between variability of hypothesized drivers and coccolithophore chlorophyll a concentrations at the BATS site. Increasing DIC, specifically the bicarbonate ion (HCO3–) fraction, showed a strong positive correlation with pigments from coccolithophores, explaining a significant fraction of the coccolithophore pigment variability. DIC in the upper mixed layer at BATS has been increasing steadily over the past several decades from absorption of anthropogenic CO2 (Fig. 2; 23) and coccolithophores may be responding to this. But how does extra carbon in the water explain the increases in coccolithophore populations?
Environmental controls on coccolithophore growth
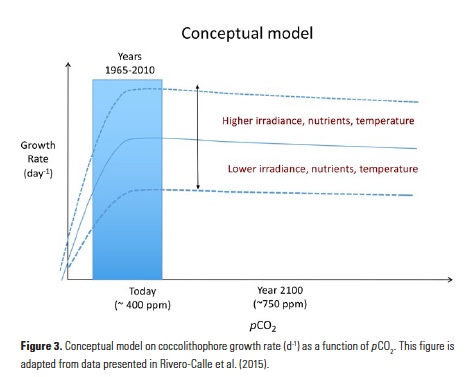
A few studies have shown that, in contrast to most other phytoplankton, coccolithophore photosynthesis (specifically, the widespread coccolithophore species Emiliania huxleyi) can be carbon-limited at today’s CO2 levels (e.g., 14, 24). This suggests that increases in surface DIC (e.g., due to the uptake of anthropogenic CO2) may alleviate growth limitation of coccolithophores. By reducing the amount of energy spent on carbon concentrating mechanisms, coccolithophores may invest in other metabolic processes such as growth, PIC or POC production. This explains why a relatively small increase in DIC could increase coccolithophore competitive ability, especially in oligotrophic environments where phytoplankton are routinely in competition for scarce nutrients. Rivero-Calle and co-authors compiled numerous published laboratory studies that assessed coccolithophore growth rates as a function of pCO2. The compilation, which included several species and strains of coccolithophores, showed that there is a quasi-hyperbolic increase in coccolithophore growth rates as pCO2 increases (Fig. 3). The range of local pCO2 concentrations in the subpolar/temperate North Atlantic from 1965 to 2010 (~175 to 435 ppm) spanned the pCO2 levels over which there is a substantial increase in published coccolithophore growth rates (Fig. 3). Growth rates tend to stabilize at ~500 ppmCO2, indicating that coccolithophore populations may continue to respond positively to increasing CO2 for the next few decades.
Other environmental factors (e.g., temperature, light, and available nutrients) may also impact and modulate coccolithophore growth rates, resulting in a net neutral or net negative impact in spite of increasing atmospheric (marine) CO2 (DIC) concentrations (see conceptual model, Fig. 3). For example, severe nutrient limitation in the subtropics may cause coccolithophores to be outcompeted by smaller marine cyanobacteria. In the subpolar North Atlantic, nutrients are more plentiful than in the subtropics, but Earth system models have predicted that climatic warming in this region may result in increased water column stratification (25). Under these stratified low-nutrient conditions, smaller phytoplankton such as coccolithophores could become more prevalent at the expense of larger phytoplankton such as diatoms (26, 27). However, if nutrient concentrations decline to the point at which they become the limiting factor for growth, then coccolithophore populations will also be negatively affected. Furthermore, the associated drop in pH from CO2 dissolving into the upper mixed layer can eventually be detrimental to coccolithophore growth and calcification. Specifically, pH values below 7.7 negatively affected the coccolithophore Emiliania huxleyi in laboratory experiments (14), though most oceanic regions will not show such a low pH any time in the near future. In short, anthropogenic CO2 entering the ocean may allow coccolithophores a competitive edge in the near future in some regions such as the North Atlantic, but other compounding influences from anthropogenic climate change such as severe nutrient limitation or ocean acidification are also important to consider, particularly in the oligotrophic gyres.
Open questions and future directions
While recent work has provided new insight into the impact of several environmental factors (irradiance, nutrients, temperature, pH, DIC) on coccolithophores, many questions remain. Among these, the vertical distribution of coccolithophore communities, grazing rates, and viral infection on coccolithophores, and species-specific responses to environmental change are relatively unexplored areas of research. For instance, some studies have shown species-specific and even strain-specific variability in the response of coccolithophores to CO2 (28, 29), but how various coccolithophore species respond to nutrient or light limitation is relatively unknown. Due to its cosmopolitan distribution and ability to grow relatively easily in the lab, E. huxleyi, has become the “lab rat” species. However, it may not be the most important calcite producer globally (30), nor the most representative of the coccolithophore group as a whole. As part of its peculiarities, E. huxleyi can both produce several layers of coccoliths and also exhibit a naked form without coccoliths, posing questions about the importance of non-calcified forms in the projected acidified oceans and about the role of calcification per se (31). Indeed, the fundamental question of why coccolithophores calcify is still unresolved and may vary between species (5, 32). In addition, while we recognize that some zooplankton groups graze on coccolithophores (coccoliths have been found in pelagic tintinnid ciliates (33), as well as copepod guts and fecal pellets (34-36)), little is known about predation rates or specificity in natural populations. Finally, we know that viruses can also cause bloom termination and that E. huxleyi can induce coccolith detachment to avoid viral invasion (37); however, there are still many unknowns related to bloom dynamics. Until we understand what drives coccolithophore calcification and variations in growth and mortality rates, we will have an incomplete picture of the role that coccolithophores play in marine ecology and the carbon cycle.
Krumhardt et al (2016) and Rivero-Calle et al (2016) both arrive at a simple conclusion: Coccolithophore presence in the North Atlantic is increasing. The common denominators in this equation are increasing global CO2 levels and increasing global surface temperatures. Therefore, even given regional oceanic variability in environmental drivers, we might expect to see similar trends in coccolithophore abundance in other regions. Given coccolithophores’ positive response to increasing anthropogenic CO2 and temperature, as well as general fitness under conditions that may be more prevalent in the future ocean, coccolithophores may become an even bigger player in the marine carbon cycle, which may have unexpected consequences.
Authors
Kristen Krumhardt (University of Colorado Boulder)
Sara Rivero-Calle (University of Southern California, Los Angeles)
Acknowledgments
We would like to thank co-authors on the Rivero-Calle et al. (2015) and Krumhardt et al. (2016) studies for their contributions to the research described. We also would like to thank Nikki Lovenduski and Naomi Levine for helpful comments in composing this OCB Newsletter piece. Many thanks to APL, NSF, NOAA and NASA for funding, and SAHFOS, ICOADS, the BATS research group, and NASA for their long-term data and making it freely available.
References
1. C. L. Sabine et al., Science 305 (2004).
2. Feely et al., Science 305, 362-366 (2004).
3. J. C. Orr et al., Nature 437, 681-686 (2005).
4. K. J. Kroeker et al., Global Change Biology 19, 1884-1896 (2013).
5. B. Rost, U. Riebesell, In Coccolithophores: from Molecular Processes to Global Impact, 99-125 (2004).
6. U. Riebesell et al., Nature 407, 364-367 (2000).
7. C. Klaas, D. E. Archer, Glob.Biogeochem. Cycles 16, (2002).
8. M. D. Keller, W. K. Bellows, R. R. L. Guillard, Acs Symposium Series 393, 167-182 (1989).
9. S. M. Vallina, R. Simo, Science 315, 506-508 (2007).
10. T. Tyrrell, A. Merico, In H. R. Thierstein, J. R. Young, Eds.,Coccolithophores: from Molecular Processes to Global Impact (2004),pp. 75-97.
11. W. M. Balch, In Coccolithophores: from Molecular Processes to Global Impact, 165-190 (2004).
12. M. D. Iglesias-Rodriguez et al., Science 320, 336-340 (2008).
13. S. Sett et al., Plos One 9, (2014).
14. L. T. Bach et al., New Phytol. 199, 121-134 (2013).
15. L. Schluter et al., Nature Clim. Change 4, 1024-1030 (2014).
16. S. Rivero-Calle, A. Gnanadesikan, C. E. Del Castillo, W. M.Balch, S. D. Guikema, Science 350, 1533-1537 (2015).
17. K. M. Krumhardt, N. S. Lovenduski, N. M. Freeman, N. R.Bates, Biogeosci. 13, 1163-1177 (2016).
18. H. Liu et al., Proc. Nat. Acad. Sci. 106, 12803-12808 (2009).
19. Y. Dandonneau, Y. Montel, J. Blanchot, J. Giraudeau, J. Neveux, Deep-Sea Res. Part I-Oceanographic Research Papers 53, 689-712(2006).
20. M. J. Behrenfeld, P. G. Falkowski, Limnol. Oceanogr. 42, 1-20(1997).
21. H. R. Gordon et al. (Geochemical Research Letters, 2001), vol.28, pp. 1587-1590.
22. W. M. Balch, H. R. Gordon, B. C. Bowler, D. T. Drapeau, E. S.
Booth, J. Geophys. Res.Oceans 110 (2005).
23. N. R. Bates et al., Oceanogr. 27, 126-141 (2014).
24. B. Rost, U. Riebesell, S. Burkhardt, D. Sultemeyer, Limnol.Oceanogr. 48, 55-67 (2003).
25. A. Cabr., I. Marinov, S. Leung, Clim.Dyn. 1-28 (2014).
26. L. Bopp, O. Aumont, P. Cadule, S. Alvain, M. Gehlen, Geophys.Res. Lett. 32, (2005).
27. I. Marinov, S. C. Doney, I. D. Lima, Biogeosci. 7, 3941-3959 (2010).
28. Langer et al., Geochem. Geophys. Geosys. 7, (2006).
29. Langer, G. Nehrke, I. Probert, J. Ly, P. Ziveri, Biogeosci. 6, 2637-2646 (2009).
30. C. J. Daniels et al., Marine Ecol. Prog. Ser. 555, 29-47 (2016).
31. M. N. Muller, T. W. Trull, G. M. Hallegraeff, Marine Ecol. Prog.Ser. 531, 81-90 (2015).
32. F. M. Monteiro et al., Science Advances 2, (2016).
33. J. Henjes, P. Assmy, Protist 159, 239-250 (2008).
34. S. Honjo, M. R. Roman, J. Marine Res. 36, 45-57 (1978).
35. R. P. Harris, Marine Biol. 119, 431-439 (1994).
36. J. D. Milliman et al., Deep Sea Res. Part I: Oceanographic Research Papers 46, 1653-1669 (1999).
37. M. Frada, I. Probert, M. J. Allen, W. H. Wilson, C. de Vargas, Proc. Nat. Acad. Sci. 105, 15944-15949 (2008).